InVivoPlus anti-mouse CD4
| Clone | GK1.5 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalog # | BP0003-1 | ||||||||||
| Category | InVivoPlus Antibodies | ||||||||||
| Price |
|
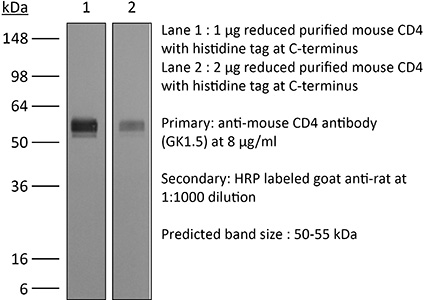
The GK1.5 monoclonal antibody reacts with the mouse CD4. The CD4 antigen is a 55 kDa cell surface type I membrane glycoprotein belonging to the immunoglobulin superfamily. CD4 acts as a co-receptor which in cooperation with the T cell receptor (TCR) interacts with class II MHC molecules displayed by antigen presenting cells (APC). CD4 is expressed by the majority of thymocytes, most helper T cells, a subset of NK-T cells and weakly by dendritic cells and macrophages. CD4 plays an important role in the development of T cells and is required for mature T cells to function optimally. The GK1.5 antibody has been shown to compete with clones YTS 177 and YTS 191 for CD4 binding.
| Isotype | Rat IgG2b, κ |
| Recommended Isotype Control(s) | InVivoPlus™ rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure™ pH 6.5 Dilution Buffer |
| Immunogen | Mouse CTL clone V4 |
| Reported Applications |
|
| Formulation |
|
| Endotoxin |
|
| Aggregation |
|
| Purity |
|
| Sterility | 0.2 μM filtered |
| Production | Purified from tissue culture supernatant in an animal free facility |
| Purification | Protein G |
| RRID | AB_1107636 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Test Results |
|
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
Balogh, K. N., et al. (2018). “Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses.” PLoS One 13(6): e0197702. PubMed
The Macrophage Migration Inhibitory Factor (MIF) is an inflammatory cytokine that is overexpressed in a number of cancer types, with increased MIF expression often correlating with tumor aggressiveness and poor patient outcomes. In this study, we aimed to better understand the link between primary tumor expression of MIF and increased tumor growth. Using the MMTV-PyMT murine model of breast cancer, we observed that elevated MIF expression promoted tumor appearance and growth. Supporting this, we confirmed our previous observation that higher MIF expression supported tumor growth in the 4T1 murine model of breast cancer. We subsequently discovered that loss of MIF expression in 4T1 cells led to decreased cell numbers and increased apoptosis in vitro under reduced serum culture conditions. We hypothesized that this increase in cell death would promote detection by the host immune system in vivo, which could explain the observed impairment in tumor growth. Supporting this, we demonstrated that loss of MIF expression in the primary tumor led to an increased abundance of intra-tumoral IFNgamma-producing CD4+ and CD8+ T cells, and that depletion of T cells from mice bearing MIF-deficient tumors restored growth to the level of MIF-expressing tumors. Furthermore, we found that MIF depletion from the tumor cells resulted in greater numbers of activated intra-tumoral dendritic cells (DCs). Lastly, we demonstrated that loss of MIF expression led to a robust induction of a specialized form of cell death, immunogenic cell death (ICD), in vitro. Together, our data suggests a model in which MIF expression in the primary tumor dampens the anti-tumor immune response, promoting tumor growth.
Budda, S. A. and L. A. Zenewicz (2018). “IL-22 deficiency increases CD4 T cell responses to mucosal immunization.” Vaccine 36(25): 3694-3700. PubMed
Mucosal vaccines are a promising platform for combatting infectious diseases for which we still lack effective preventative measures. Optimizing these vaccines to generate the best protective immune responses with the least complicated immunization regimen is imperative. Mucosal barriers are the first line of defense against many pathogens and, as such, we looked to their biology for strategies to improve vaccine delivery. Interleukin-22 (IL-22) is a key cytokine in both healthy and inflamed mucosal tissues. IL-22 promotes epithelial cell proliferation and inhibits apoptosis, upregulates mucin and antimicrobial peptides, all of which promote mucosal barrier integrity. In this study, we find that IL-22 impairs the development of a T cell response during mucosal immunization. Compared to wild-type control mice, IL-22 deficient mice had increased antigen-specific CD4 T cell responses to intrarectal immunization using a protein and cholera toxin adjuvant vaccine. When immunized systemically with the same protein antigen adsorbed to alum, no differences in the CD4 T cell response between wild-type and IL-22 deficient mice were detected. This suggests that transiently inhibiting IL-22 during mucosal vaccination could enhance T cell responses. The broad-applicability of this proposed approach would allow for improvement of many existing mucosal vaccine regimens and have positive implications in the development of more efficacious mucosal vaccines.
Moynihan, K. D., et al. (2016). “Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses.” Nat Med. doi: 10.1038/nm.4200. PubMed
Checkpoint blockade with antibodies specific for cytotoxic T lymphocyte-associated protein (CTLA)-4 or programmed cell death 1 (PDCD1; also known as PD-1) elicits durable tumor regression in metastatic cancer, but these dramatic responses are confined to a minority of patients. This suboptimal outcome is probably due in part to the complex network of immunosuppressive pathways present in advanced tumors, which are unlikely to be overcome by intervention at a single signaling checkpoint. Here we describe a combination immunotherapy that recruits a variety of innate and adaptive immune cells to eliminate large tumor burdens in syngeneic tumor models and a genetically engineered mouse model of melanoma; to our knowledge tumors of this size have not previously been curable by treatments relying on endogenous immunity. Maximal antitumor efficacy required four components: a tumor-antigen-targeting antibody, a recombinant interleukin-2 with an extended half-life, anti-PD-1 and a powerful T cell vaccine. Depletion experiments revealed that CD8+ T cells, cross-presenting dendritic cells and several other innate immune cell subsets were required for tumor regression. Effective treatment induced infiltration of immune cells and production of inflammatory cytokines in the tumor, enhanced antibody-mediated tumor antigen uptake and promoted antigen spreading. These results demonstrate the capacity of an elicited endogenous immune response to destroy large, established tumors and elucidate essential characteristics of combination immunotherapies that are capable of curing a majority of tumors in experimental settings typically viewed as intractable.
Christensen, A. D., et al. (2015). “Depletion of regulatory T cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T cells.” Clin Exp Immunol 179(3): 485-499. PubMed
Regulatory T cells (Tregs ) are known to play an immunosuppressive role in the response of contact hypersensitivity (CHS), but neither the dynamics of Tregs during the CHS response nor the exaggerated inflammatory response after depletion of Tregs has been characterized in detail. In this study we show that the number of Tregs in the challenged tissue peak at the same time as the ear-swelling reaches its maximum on day 1 after challenge, whereas the number of Tregs in the draining lymph nodes peaks at day 2. As expected, depletion of Tregs by injection of a monoclonal antibody to CD25 prior to sensitization led to a prolonged and sustained inflammatory response which was dependent upon CD8 T cells, and co-stimulatory blockade with cytotoxic T lymphocyte antigen-4-immunoglobulin (CTLA-4-Ig) suppressed the exaggerated inflammation. In contrast, blockade of the interleukin (IL)-10-receptor (IL-10R) did not further increase the exaggerated inflammatory response in the Treg -depleted mice. In the absence of Tregs , the response changed from a mainly acute reaction with heavy infiltration of neutrophils to a sustained response with more chronic characteristics (fewer neutrophils and dominated by macrophages). Furthermore, depletion of Tregs enhanced the release of cytokines and chemokines locally in the inflamed ear and augmented serum levels of the systemic inflammatory mediators serum amyloid (SAP) and haptoglobin early in the response.
Evans, E. E., et al. (2015). “Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies.” Cancer Immunol Res 3(6): 689-701. PubMed
Semaphorin 4D (SEMA4D, CD100) and its receptor plexin-B1 (PLXNB1) are broadly expressed in murine and human tumors, and their expression has been shown to correlate with invasive disease in several human tumors. SEMA4D normally functions to regulate the motility and differentiation of multiple cell types, including those of the immune, vascular, and nervous systems. In the setting of cancer, SEMA4D-PLXNB1 interactions have been reported to affect vascular stabilization and transactivation of ERBB2, but effects on immune-cell trafficking in the tumor microenvironment (TME) have not been investigated. We describe a novel immunomodulatory function of SEMA4D, whereby strong expression of SEMA4D at the invasive margins of actively growing tumors influences the infiltration and distribution of leukocytes in the TME. Antibody neutralization of SEMA4D disrupts this gradient of expression, enhances recruitment of activated monocytes and lymphocytes into the tumor, and shifts the balance of cells and cytokines toward a proinflammatory and antitumor milieu within the TME. This orchestrated change in the tumor architecture was associated with durable tumor rejection in murine Colon26 and ERBB2(+) mammary carcinoma models. The immunomodulatory activity of anti-SEMA4D antibody can be enhanced by combination with other immunotherapies, including immune checkpoint inhibition and chemotherapy. Strikingly, the combination of anti-SEMA4D antibody with antibody to CTLA-4 acts synergistically to promote complete tumor rejection and survival. Inhibition of SEMA4D represents a novel mechanism and therapeutic strategy to promote functional immune infiltration into the TME and inhibit tumor progression.
Guo, L., et al. (2015). “Innate immunological function of TH2 cells in vivo.” Nat Immunol 16(10): 1051-1059. PubMed
Type 2 helper T cells (TH2 cells) produce interleukin 13 (IL-13) when stimulated by papain or house dust mite extract (HDM) and induce eosinophilic inflammation. This innate response is dependent on IL-33 but not T cell antigen receptors (TCRs). While type 2 innate lymphoid cells (ILC2 cells) are the dominant innate producers of IL-13 in naive mice, we found here that helminth-infected mice had more TH2 cells compared to uninfected mice, and thes e cells became major mediators of innate type 2 responses. TH2 cells made important contributions to HDM-induced antigen-nonspecific eosinophilic inflammation and protected mice recovering from infection with Ascaris suum against subsequent infection with the phylogenetically distant nematode Nippostrongylus brasiliensis. Our findings reveal a previously unappreciated role for effector TH2 cells during TCR-independent innate-like immune responses.
Kim, J., et al. (2015). “Memory programming in CD8(+) T-cell differentiation is intrinsic and is not determined by CD4 help.” Nat Commun 6: 7994. PubMed
CD8(+) T cells activated without CD4(+) T-cell help are impaired in memory expansion. To understand the underlying cellular mechanism, here we track the dynamics of helper-deficient CD8(+) T-cell response to a minor histocompatibility antigen by phenotypic and in vivo imaging analyses. Helper-deficient CD8(+) T cells show reduced burst expansion, rapid peripheral egress, delayed antigen clearance and continuous activation, and are eventually exhausted. Contrary to the general consensus that CD4 help encodes memory programmes in CD8(+) T cells and helper-deficient CD8(+) T cells are abortive, these cells can differentiate into effectors and memory precursors. Importantly, accelerating antigen clearance or simply increasing the burst effector size enables generation of memory cells by CD8(+) T cells, regardless of CD4 help. These results suggest that the memory programme is CD8(+) T-cell-intrinsic, and provide insight into the role of CD4 help in CD8(+) T-cell responses.
Liu, G., et al. (2015). “IL-27 Signaling Is Crucial for Survival of Mice Infected with African Trypanosomes via Preventing Lethal Effects of CD4+ T Cells and IFN-gamma.” PLoS Pathog 11(7): e1005065. PubMed
African trypanosomes are extracellular protozoan parasites causing a chronic debilitating disease associated with a persistent inflammatory response. Maintaining the balance of the inflammatory response via downregulation of activation of M1-type myeloid cells was previously shown to be crucial to allow prolonged survival. Here we demonstrate that infection with African trypanosomes of IL-27 receptor-deficient (IL-27R-/-) mice results in severe liver immunopathology and dramatically reduced survival as compared to wild-type mice. This coincides with the development of an exacerbated Th1-mediated immune response with overactivation of CD4+ T cells and strongly enhanced production of inflammatory cytokines including IFN-gamma. What is important is that IL-10 production was not impaired in infected IL-27R-/- mice. Depletion of CD4+ T cells in infected IL-27R-/- mice resulted in a dramatically reduced production of IFN-gamma, preventing the early mortality of infected IL-27R-/- mice. This was accompanied by a significantly reduced inflammatory response and a major amelioration of liver pathology. These results could be mimicked by treating IL-27R-/- mice with a neutralizing anti-IFN-gamma antibody. Thus, our data identify IL-27 signaling as a novel pathway to prevent early mortality via inhibiting hyperactivation of CD4+ Th1 cells and their excessive secretion of IFN-gamma during infection with African trypanosomes. These data are the first to demonstrate the essential role of IL-27 signaling in regulating immune responses to extracellular protozoan infections.
Vanpouille-Box, C., et al. (2015). “TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity.” Cancer Res 75(11): 2232-2242. PubMed
T cells directed to endogenous tumor antigens are powerful mediators of tumor regression. Recent immunotherapy advances have identified effective interventions to unleash tumor-specific T-cell activity in patients who naturally develop them. Eliciting T-cell responses to a patient’s individual tumor remains a major challenge. Radiation therapy can induce immune responses to model antigens expressed by tumors, but it remains unclear whether it can effectively prime T cells specific for endogenous antigens expressed by poorly immunogenic tumors. We hypothesized that TGFbeta activity is a major obstacle hindering the ability of radiation to generate an in situ tumor vaccine. Here, we show that antibody-mediated TGFbeta neutralization during radiation therapy effectively generates CD8(+) T-cell responses to multiple endogenous tumor antigens in poorly immunogenic mouse carcinomas. Generated T cells were effective at causing regression of irradiated tumors and nonirradiated lung metastases or synchronous tumors (abscopal effect). Gene signatures associated with IFNgamma and immune-mediated rejection were detected in tumors treated with radiation therapy and TGFbeta blockade in combination but not as single agents. Upregulation of programmed death (PD) ligand-1 and -2 in neoplastic and myeloid cells and PD-1 on intratumoral T cells limited tumor rejection, resulting in rapid recurrence. Addition of anti-PD-1 antibodies extended survival achieved with radiation and TGFbeta blockade. Thus, TGFbeta is a fundamental regulator of radiation therapy’s ability to generate an in situ tumor vaccine. The combination of local radiation therapy with TGFbeta neutralization offers a novel individualized strategy for vaccinating patients against their tumors.
Zander, R. A., et al. (2015). “PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity.” Cell Host Microbe 17(5): 628-641. PubMed
The differentiation and protective capacity of Plasmodium-specific T cells are regulated by both positive and negative signals during malaria, but the molecular and cellular details remain poorly defined. Here we show that malaria patients and Plasmodium-infected rodents exhibit atypical expression of the co-stimulatory receptor OX40 on CD4 T cells and that therapeutic enhancement of OX40 signaling enhances helper CD4 T cell activity, humoral immunity, and parasite clearance in rodents. However, these beneficial effects of OX40 signaling are abrogated following coordinate blockade of PD-1 co-inhibitory pathways, which are also upregulated during malaria and associated with elevated parasitemia. Co-administration of biologics blocking PD-1 and promoting OX40 signaling induces excessive interferon-gamma that directly limits helper T cell-mediated support of humoral immunity and decreases parasite control. Our results show that targeting OX40 can enhance Plasmodium control and that crosstalk between co-inhibitory and co-stimulatory pathways in pathogen-specific CD4 T cells can impact pathogen clearance.
Church, S. E., et al. (2014). “Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells.” Eur J Immunol 44(1): 69-79. PubMed
Immunotherapies that augment antitumor T cells have had recent success for treating patients with cancer. Here we examined whether tumor-specific CD4(+) T cells enhance CD8(+) T-cell adoptive immunotherapy in a lymphopenic environment. Our model employed physiological doses of tyrosinase-related protein 1-specific CD4(+) transgenic T cells-CD4(+) T cells and pmel-CD8(+) T cells that when transferred individually were subtherapeutic; however, when transferred together provided significant (p
Krupnick, A. S., et al. (2014). “Central memory CD8+ T lymphocytes mediate lung allograft acceptance.” J Clin Invest 124(3): 1130-1143. PubMed
Memory T lymphocytes are commonly viewed as a major barrier for long-term survival of organ allografts and are thought to accelerate rejection responses due to their rapid infiltration into allografts, low threshold for activation, and ability to produce inflammatory mediators. Because memory T cells are usually associated with rejection, preclinical protocols have been developed to target this population in transplant recipients. Here, using a murine model, we found that costimulatory blockade-mediated lung allograft acceptance depended on the rapid infiltration of the graft by central memory CD8+ T cells (CD44(hi)CD62L(hi)CCR7+). Chemokine receptor signaling and alloantigen recognition were required for trafficking of these memory T cells to lung allografts. Intravital 2-photon imaging revealed that CCR7 expression on CD8+ T cells was critical for formation of stable synapses with antigen-presenting cells, resulting in IFN-gamma production, which induced NO and downregulated alloimmune responses. Thus, we describe a critical role for CD8+ central memory T cells in lung allograft acceptance and highlight the need for tailored approaches for tolerance induction in the lung.
Uddin, M. N., et al. (2014). “TNF-alpha-dependent hematopoiesis following Bcl11b deletion in T cells restricts metastatic melanoma.” J Immunol 192(4): 1946-1953. PubMed
Using several tumor models, we demonstrate that mice deficient in Bcl11b in T cells, although having reduced numbers of T cells in the peripheral lymphoid organs, developed significantly less tumors compared with wild-type mice. Bcl11b(-/-) CD4(+) T cells, with elevated TNF-alpha levels, but not the Bcl11b(-/-) CD8(+) T cells, were required for the reduced tumor burden, as were NK1.1(+) cells, found in increased numbers in Bcl11b(F/F)/CD4-Cre mice. Among NK1.1(+) cells, the NK cell population was predominant in number and was the only population displaying elevated granzyme B levels and increased degranulation, although not increased proliferation. Although the number of myeloid-derived suppressor cells was increased in the lungs with metastatic tumors of Bcl11b(F/F)/CD4-Cre mice, their arginase-1 levels were severely reduced. The increase in NK cell and myeloid-derived suppressor cell numbers was associated with increased bone marrow and splenic hematopoiesis. Finally, the reduced tumor burden, increased numbers of NK cells in the lung, and increased hematopoiesis in Bcl11b(F/F)/CD4-Cre mice were all dependent on TNF-alpha. Moreover, TNF-alpha treatment of wild-type mice also reduced the tumor burden and increased hematopoiesis and the numbers and activity of NK cells in the lung. In vitro treatment with TNF-alpha of lineage-negative hematopoietic progenitors increased NK and myeloid differentiation, further supporting a role of TNF-alpha in promoting hematopoiesis. These studies reveal a novel role for TNF-alpha in the antitumor immune response, specifically in stimulating hematopoiesis and increasing the numbers and activity of NK cells.
Xin, L., et al. (2014). “Commensal microbes drive intestinal inflammation by IL-17-producing CD4+ T cells through ICOSL and OX40L costimulation in the absence of B7-1 and B7-2.” Proc Natl Acad Sci U S A 111(29): 10672-10677. PubMed
The costimulatory B7-1 (CD80)/B7-2 (CD86) molecules, along with T-cell receptor stimulation, together facilitate T-cell activation. This explains why in vivo B7 costimulation neutralization efficiently silences a variety of human autoimmune disorders. Paradoxically, however, B7 blockade also potently moderates accumulation of immune-suppressive regulatory T cells (Tregs) essential for protection against multiorgan systemic autoimmunity. Here we show that B7 deprivation in mice overrides the necessity for Tregs in averting systemic autoimmunity and inflammation in extraintestinal tissues, whereas peripherally induced Tregs retained in the absence of B7 selectively mitigate intestinal inflammation caused by Th17 effector CD4(+) T cells. The need for additional immune suppression in the intestine reflects commensal microbe-driven T-cell activation through the accessory costimulation molecules ICOSL and OX40L. Eradication of commensal enteric bacteria mitigates intestinal inflammation and IL-17 production triggered by Treg depletion in B7-deficient mice, whereas re-establishing intestinal colonization with Candida albicans primes expansion of Th17 cells with commensal specificity. Thus, neutralizing B7 costimulation uncovers an essential role for Tregs in selectively averting intestinal inflammation by Th17 CD4(+) T cells with commensal microbe specificity.
Dai, M., et al. (2013). “Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation.” J Immunother 36(4): 248-257. PubMed
Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
Hervieu, A., et al. (2013). “Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth.” J Invest Dermatol 133(2): 499-508. PubMed
Dacarbazine (DTIC) is a cytotoxic drug widely used for melanoma treatment. However, the putative contribution of anticancer immune responses in the efficacy of DTIC has not been evaluated. By testing how DTIC affects host immune responses to cancer in a mouse model of melanoma, we unexpectedly found that both natural killer (NK) and CD8(+) T cells were indispensable for DTIC therapeutic effect. Although DTIC did not directly affect immune cells, it triggered the upregulation of NKG2D ligands on tumor cells, leading to NK cell activation and IFNgamma secretion in mice and humans. NK cell-derived IFNgamma subsequently favored upregulation of major histocompatibility complex class I molecules on tumor cells, rendering them sensitive to cytotoxic CD8(+) T cells. Accordingly, DTIC markedly enhanced cytotoxic T lymphocyte antigen 4 inhibition efficacy in vivo in an NK-dependent manner. These results underscore the immunogenic properties of DTIC and provide a rationale to combine DTIC with immunotherapeutic agents that relieve immunosuppression in vivo.
Butler, N. S., et al. (2012). “Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection.” Nat Immunol 13(2): 188-195. PubMed
Infection of erythrocytes with Plasmodium species induces clinical malaria. Parasite-specific CD4(+) T cells correlate with lower parasite burdens and severity of human malaria and are needed to control blood-stage infection in mice. However, the characteristics of CD4(+) T cells that determine protection or parasite persistence remain unknown. Here we show that infection of humans with Plasmodium falciparum resulted in higher expression of the inhibitory receptor PD-1 associated with T cell dysfunction. In vivo blockade of the PD-1 ligand PD-L1 and the inhibitory receptor LAG-3 restored CD4(+) T cell function, amplified the number of follicular helper T cells and germinal-center B cells and plasmablasts, enhanced protective antibodies and rapidly cleared blood-stage malaria in mice. Thus, chronic malaria drives specific T cell dysfunction, and proper function can be restored by inhibitory therapies to enhance parasite control.
Krieg, C., et al. (2010). “Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells.” Proc Natl Acad Sci U S A 107(26): 11906-11911. PubMed
IL-2 immunotherapy is an attractive treatment option for certain metastatic cancers. However, administration of IL-2 to patients can lead, by ill-defined mechanisms, to toxic adverse effects including severe pulmonary edema. Here, we show that IL-2-induced pulmonary edema is caused by direct interaction of IL-2 with functional IL-2 receptors (IL-2R) on lung endothelial cells in vivo. Treatment of mice with high-dose IL-2 led to efficient expansion of effector immune cells expressing high levels of IL-2Rbetagamma, including CD8(+) T cells and natural killer cells, which resulted in a considerable antitumor response against s.c. and pulmonary B16 melanoma nodules. However, high-dose IL-2 treatment also affected immune cell lineage marker-negative CD31(+) pulmonary endothelial cells via binding to functional alphabetagamma IL-2Rs, expressed at low to intermediate levels on these cells, thus causing pulmonary edema. Notably, IL-2-mediated pulmonary edema was abrogated by a blocking antibody to IL-2Ralpha (CD25), genetic disruption of CD25, or the use of IL-2Rbetagamma-directed IL-2/anti-IL-2 antibody complexes, thereby interfering with IL-2 binding to IL-2Ralphabetagamma(+) pulmonary endothelial cells. Moreover, IL-2/anti-IL-2 antibody complexes led to vigorous activation of IL-2Rbetagamma(+) effector immune cells, which generated a dramatic antitumor response. Thus, IL-2/anti-IL-2 antibody complexes might improve current strategies of IL-2-based tumor immunotherapy.
하단영역
(ZIP code : 07532) 902, 9F Hanwha bizmetro 1-cha, 551-17 YangcheonRo,
Gayang-dong, Gangseo-Gu, Seoul, Korea
TEL : 02-2013-8240
FAX : 02-2013-8243
Email : bioxcell@bioxcell.co.kr
Copyright by 2020 BioXCell Korea. All Rights Reserved.
- BioXCell Korea
- Opening hours
- Monday - Friday 9AM - 6PM
- Follow Us
-



